.svg)





What cellular senescence actually is at a molecular level
Cellular senescence is a state in which a cell permanently exits the cell cycle and stops dividing, but remains metabolically active and resistant to programmed cell death. Unlike quiescent cells, which can re-enter the cell cycle when conditions change, senescent cells are locked in place. They don't replicate, but they don't quietly fade away either.
What makes senescent cells particularly consequential is not their inactivity but their activity. They secrete a complex mixture of pro-inflammatory cytokines, growth factors, proteases, and other signaling molecules collectively known as the senescence-associated secretory phenotype (SASP). This secretory profile can alter the behavior of neighboring cells, degrade the extracellular matrix, and recruit immune cells to the site.
In the short term, this is useful. SASP signals help coordinate wound healing, prevent damaged cells from becoming cancerous, and facilitate tissue remodeling during development. But when senescent cells accumulate and persist, the chronic inflammatory signaling becomes a liability. The molecular machinery that enforces senescence centers on two major tumor suppressor pathways: p53 and p16 (mechanisms and functions of cellular senescence).
When a cell experiences significant stress, DNA damage response pathways activate p53, which can trigger cell cycle arrest, DNA repair, or apoptosis. If the damage is irreparable but the cell survives, p53 can induce senescence. The p16 pathway, regulated by the INK4a/ARF locus, provides a parallel route into senescence, often activated by telomere dysfunction or oncogene activation. Once either pathway is engaged persistently, the cell enters a stable arrest that is extremely difficult to reverse.
How cells enter senescence through DNA damage and other triggers
Cellular senescence can be triggered by a wide range of stressors, but they converge on a common theme: genomic or cellular damage that the cell cannot fully repair. The primary triggers include:
- DNA damage and telomere attrition result from oxidative stress, ionizing radiation, chemotherapy, or replication errors that activate chronic DNA damage responses.
- Oncogene activation paradoxically induces senescence as a tumor suppressor mechanism, preventing abnormal proliferative signals from driving uncontrolled cell division.
- Mitochondrial dysfunction generates excessive reactive oxygen species that damage cellular components and release mitochondrial DNA into the cytoplasm, activating inflammatory pathways.
- Epigenetic disruption through loss of heterochromatin and DNA methylation changes activates senescence programs and contributes to age-related gene expression drift.
When DNA damage persists and cannot be repaired, the DNA damage response remains chronically activated, locking the cell into senescence. Telomeres, the protective caps at the ends of chromosomes, shorten with each cell division. When telomeres become critically short, they are recognized as DNA breaks, triggering a senescence response even in the absence of other damage. This is the basis of replicative senescence, first described by Leonard Hayflick in the 1960s.
Where cellular senescence sits within the hallmarks of aging
Cellular senescence is recognized as one of the primary hallmarks of aging. It does not act in isolation. Senescence is both a consequence of and a contributor to several other hallmarks:
- Genomic instability is both triggered by and amplified through senescence, as SASP factors damage DNA in neighboring cells, creating feedback loops.
- Telomere attrition directly induces replicative senescence when shortened telomeres lose protective function and activate DNA damage responses.
- Mitochondrial dysfunction exhibits bidirectional relationships with senescence, as impaired mitochondria trigger senescence while senescent cells show compromised mitochondrial function.
- Chronic inflammation is driven by SASP secretion of interleukin-6, interleukin-8, and tumor necrosis factor-alpha, elevating systemic inflammatory tone and accelerating tissue dysfunction.
- Stem cell exhaustion occurs when SASP factors alter the stem cell niche microenvironment, reducing regenerative capacity and tissue repair.
What drives senescent cell accumulation and why clearance fails with age
In young, healthy individuals, the immune system efficiently recognizes and eliminates senescent cells through natural killer cells and macrophages. This process prevents senescent cells from accumulating and limits their potential to cause harm. However, immune function declines with age, reducing the efficiency of senescent cell clearance.
At the same time, the rate at which new senescent cells are generated increases due to cumulative DNA damage, oxidative stress, and mitochondrial dysfunction. The result is a growing burden of senescent cells that persist in tissues, continuously secreting SASP factors and driving chronic inflammation. Senescent cells also develop mechanisms to evade immune clearance by upregulating anti-apoptotic pathways, making them resistant to signals that would normally trigger cell death.
Some senescent cells express immune checkpoint molecules that inhibit immune cell activity, further protecting themselves from elimination. This combination of increased generation, impaired clearance, and active immune evasion explains why senescent cell burden rises exponentially with age.
Why individual senescence burden varies so widely
Not everyone accumulates senescent cells at the same rate. Two individuals of the same chronological age can have vastly different senescent cell burdens, and this variation contributes to differences in biological age and healthspan. Key factors include:
- Genetic variation in DNA repair, telomere maintenance, and immune function genes influences accumulation rates and clearance efficiency.
- Cumulative stress exposure from oxidative stress, environmental toxins, ultraviolet radiation, and chronic psychological stress accelerates cellular senescence entry.
- Metabolic health status affects senescence through insulin resistance, hyperglycemia, and obesity, which promote cellular senescence via oxidative stress and mitochondrial dysfunction.
- Immune system resilience varies between individuals, with those maintaining robust natural killer cell and macrophage function better able to clear senescent cells.
What the evidence actually shows about senescence and longevity
The causal role of cellular senescence in aging has been demonstrated most convincingly in animal models. Genetic and pharmacological interventions that selectively eliminate senescent cells, a strategy known as senolytic therapy, extend healthspan and lifespan in mice. Mice engineered to clear senescent cells on demand show delayed onset of age-related pathologies including frailty, kidney dysfunction, and cardiac disease.
In humans, the evidence is more correlational but increasingly robust. Senescent cell burden, measured by markers such as p16 expression and SASP factor levels, increases with age and correlates with frailty, chronic disease burden, and mortality risk. Tissues from individuals with age-related diseases including atherosclerosis, osteoarthritis, and chronic obstructive pulmonary disease show elevated senescent cell accumulation compared to age-matched controls.
Senolytic drugs, which selectively induce apoptosis in senescent cells, are in early-stage human trials. Small pilot studies have shown that senolytics can reduce senescent cell markers, improve physical function in individuals with idiopathic pulmonary fibrosis, and reduce inflammation in patients with diabetic kidney disease. However, these are proof-of-concept studies in disease populations, not longevity trials in healthy individuals. Whether clearing senescent cells extends human lifespan remains unknown.
The challenge in translating animal data to humans is that senescent cells are not uniformly harmful. In acute settings, such as wound healing and tissue repair, senescence is beneficial. The goal is not to eliminate all senescent cells but to prevent their chronic accumulation. Timing, dosing, and patient selection for senolytic interventions remain open questions.
Measuring senescent cell burden and biological aging
Unlike many aging processes, cellular senescence can be measured, though not yet with a single simple blood test. Senescent cell burden is typically assessed through tissue biopsies, which are invasive and impractical for routine monitoring. Researchers use markers such as p16 expression, senescence-associated beta-galactosidase activity, and SASP factor secretion to identify senescent cells in tissue samples.
Circulating biomarkers offer a more accessible approach. Elevated levels of SASP factors, including interleukin-6, interleukin-8, and matrix metalloproteinase-9, in blood can reflect systemic senescent cell burden. High-sensitivity C-reactive protein, a marker of systemic inflammation, is often elevated in individuals with high senescent cell burden and correlates with biological age.
Epigenetic clocks, which estimate biological age based on DNA methylation patterns, capture the cumulative effects of cellular senescence and other aging processes. Clocks such as GrimAge and DunedinPACE integrate senescence-related changes and predict mortality risk more accurately than chronological age. While these clocks do not measure senescent cells directly, they reflect the downstream consequences of senescence accumulation, including chronic inflammation and tissue dysfunction.
Tracking inflammatory markers over time provides insight into whether senescent cell burden is increasing or stabilizing. A rising trajectory of hsCRP, interleukin-6, or other SASP-related markers suggests accelerating biological aging. Interventions that reduce inflammation, such as exercise, caloric restriction, and potentially senolytics, may slow this trajectory.
Building a data-driven view of your senescence risk
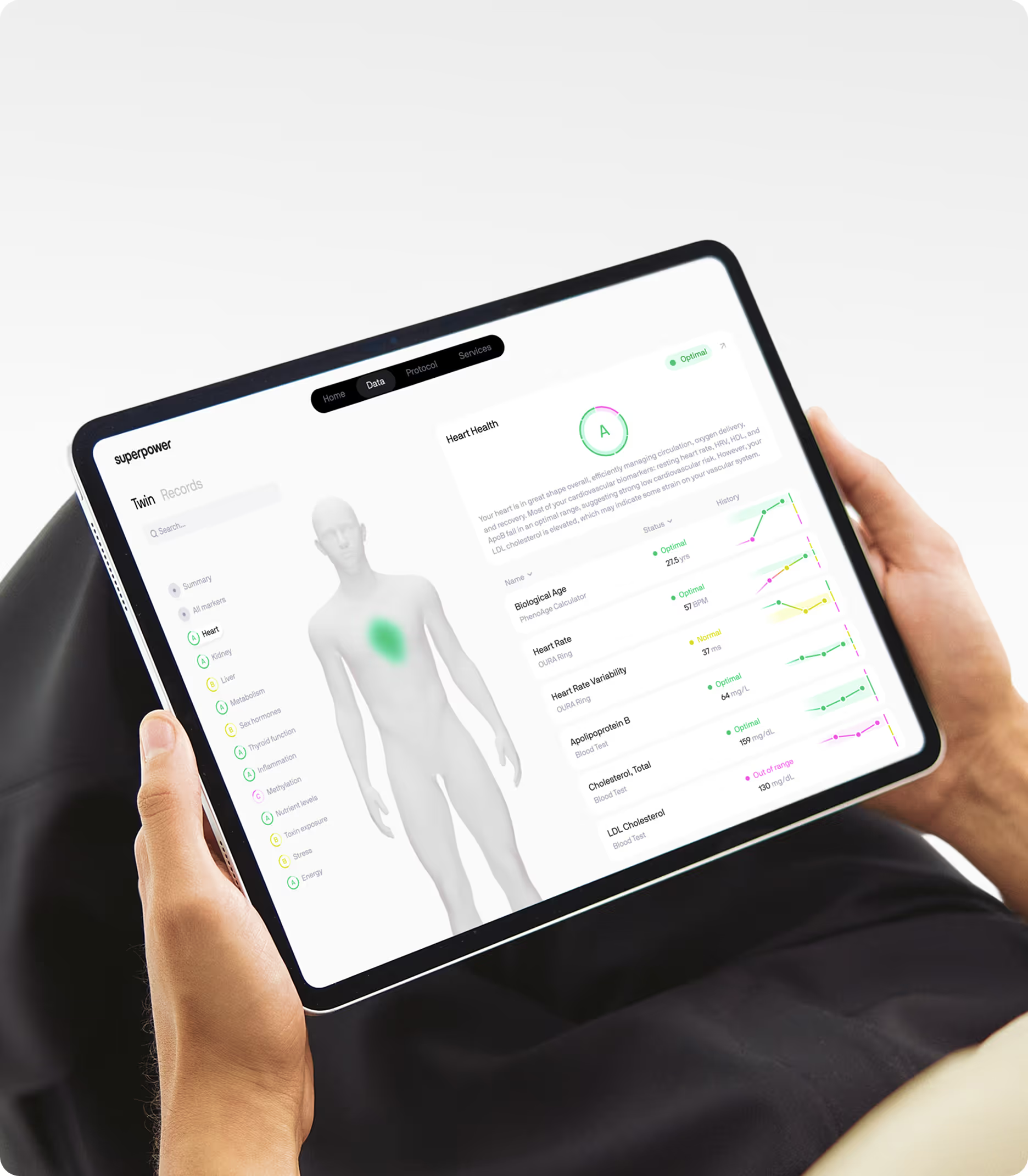
Understanding your senescent cell burden starts with measuring the markers that reflect chronic inflammation and metabolic dysfunction, the two processes most tightly linked to senescence accumulation. Superpower's 100+ biomarker panel includes high-sensitivity C-reactive protein, fasting glucose, insulin, hemoglobin A1c, and lipid markers that together provide a picture of the metabolic and inflammatory environment driving cellular aging. Tracking these markers longitudinally reveals whether your biological trajectory is accelerating or stabilizing, giving you actionable insight into how well your body is managing the cellular damage that drives senescence.



FAQs
Cellular senescence is a state in which a cell permanently exits the cell cycle and stops dividing, but remains metabolically active and resistant to apoptosis. Unlike quiescent cells that can resume dividing, senescent cells are locked in place. They are not passive — they actively secrete inflammatory cytokines, proteases, and growth factors collectively called the SASP, reshaping the tissue environment around them.
Cells enter senescence in response to irreparable stress. Primary triggers include DNA damage from oxidative stress, radiation, or replication errors; telomere attrition when chromosomal caps shorten critically; oncogene activation as a tumor suppression mechanism; mitochondrial dysfunction that increases reactive oxygen species; and epigenetic disruption. When DNA damage persists and cannot be repaired, chronic activation of the DNA damage response locks the cell into senescence.
Senescent cells secrete the SASP, a complex mixture of pro-inflammatory cytokines like IL-6 and IL-8, matrix metalloproteinases that degrade the extracellular matrix, and growth factors that alter neighboring cell behavior. SASP factors can induce senescence in adjacent healthy cells through paracrine signaling, spreading dysfunction across tissue. They also impair stem cell niches, reducing regenerative capacity and tissue repair over time.
In young individuals, natural killer cells and macrophages efficiently recognize and eliminate senescent cells. With age, immune surveillance declines while the rate of senescent cell generation increases due to cumulative DNA damage, oxidative stress, and mitochondrial dysfunction. Additionally, senescent cells develop immune evasion strategies by upregulating anti-apoptotic pathways and expressing checkpoint molecules that inhibit immune cell activity, creating an exponentially rising burden.
Senescence is protective against cancer in the short term by preventing damaged or oncogene-activated cells from proliferating. However, the SASP creates a chronic inflammatory microenvironment that can promote cancer in neighboring cells over time, and senescent cells secrete growth factors and matrix-remodeling enzymes that facilitate tumor invasion and metastasis. This dual nature means the relationship between senescence and cancer is context-dependent and timing-sensitive.
Cellular senescence is recognized as one of the primary hallmarks of aging and amplifies several others. Genomic instability and telomere attrition directly trigger senescence, while SASP factors promote further DNA damage in neighboring cells. Chronic inflammation from SASP secretion drives stem cell exhaustion, altered intercellular communication, and mitochondrial dysfunction. Senescence is both a downstream consequence of cellular damage and an upstream driver of systemic aging.
References
- National Institute on Aging. (n.d.). Does cellular senescence hold secrets for healthier aging?. https://nia.nih.gov/news/does-cellular-senescence-hold-secrets-healthier-aging
- Kumari, R., & Jat, P. (2021). Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Frontiers in cell and developmental biology, 9, 645593. https://doi.org/10.3389/fcell.2021.645593
- Ajoolabady, A., Pratico, D., Bahijri, S., Tuomilehto, J., Uversky, V. N., & Ren, J. (2025). Hallmarks of cellular senescence: biology, mechanisms, regulations. Experimental & molecular medicine, 57(7), 1482-1491. https://doi.org/10.1038/s12276-025-01480-7
- Huang, W., Hickson, L. J., Eirin, A., Kirkland, J. L., & Lerman, L. O. (2022). Cellular senescence: the good, the bad and the unknown. Nature reviews. Nephrology, 18(10), 611-627. https://doi.org/10.1038/s41581-022-00601-z
- Herranz, N., & Gil, J. (2018). Mechanisms and functions of cellular senescence. The Journal of clinical investigation, 128(4), 1238-1246. https://doi.org/10.1172/JCI95148

























.avif)
.svg)
.svg)


