.svg)





What the compression of morbidity actually means
The compression of morbidity is the idea that the period of illness and disability at the end of life can be shortened, not lengthened, even as lifespan increases. Introduced by Stanford physician James Fries in 1980, the hypothesis holds that if the age at which chronic disease and disability first appear can be postponed more rapidly than the age at death, then the total burden of lifetime illness decreases (Fries' original 1980 compression of morbidity paper). Instead of spending decades managing multiple chronic conditions, you remain functionally healthy until a brief period of decline immediately before death.
This is not about living forever. It's about changing the shape of the aging curve. In a scenario of expanding morbidity, lifespan increases but so does the number of years spent sick. Medical advances keep people alive longer, but they don't prevent the onset of disease. In compression, the onset of significant morbidity is delayed to a greater degree than mortality is postponed. The result is a shorter period of functional impairment, even if total lifespan doesn't change dramatically.
The mechanism is straightforward: if you can delay the first appearance of cardiovascular disease, diabetes, dementia, or frailty by five or ten years, and if death occurs only a few years later than it would have otherwise, you've compressed the period of illness. The key variable is not how long you live, but when chronic disease begins. Compression depends on interventions that push back disease onset faster than they extend survival.
How compression connects to the hallmarks of aging
Compression of morbidity is not a single biological process. It's the cumulative result of slowing multiple aging pathways simultaneously. The hallmarks of aging provide the mechanistic framework for understanding how compression happens at a cellular level.
Delaying the onset of chronic disease means attenuating the hallmarks that drive it. Chronic inflammation, or inflammaging, accelerates cardiovascular disease, insulin resistance, and neurodegeneration. Interventions that reduce systemic inflammatory tone, such as regular physical activity and maintenance of lean muscle mass, delay the clinical manifestation of these conditions.
Cellular senescence, the accumulation of dysfunctional cells that secrete pro-inflammatory signals, compounds this effect. Clearing senescent cells in animal models delays age-related pathologies and extends healthspan more than lifespan, a pattern consistent with compression (compression and expansion of morbidity: secular trends). Mitochondrial dysfunction drives energy deficits that manifest as fatigue, sarcopenia, and metabolic disease. Aerobic exercise stimulates mitochondrial biogenesis through PGC-1alpha activation, preserving cellular energy production and delaying functional decline.
Loss of proteostasis, the cell's ability to maintain properly folded proteins, contributes to neurodegenerative diseases like Alzheimer's and Parkinson's. Interventions that support autophagy, such as time-restricted eating or periodic fasting, enhance protein quality control and may delay the onset of these conditions. Epigenetic alterations accumulate with age and are reflected in biological age clocks like GrimAge and DunedinPACE. These clocks predict disease risk and mortality more accurately than chronological age.
Nutrient sensing and metabolic regulation
Deregulated nutrient sensing, particularly through the mTOR and insulin/IGF-1 pathways, is central to aging rate. Chronic mTOR activation from excessive caloric intake and high protein consumption accelerates cellular aging. Caloric restriction and intermittent fasting downregulate these pathways, activating AMPK and promoting autophagy. In model organisms, these interventions extend healthspan more than lifespan, compressing the period of age-related decline. In humans, the CALERIE trial demonstrated that modest caloric restriction improved metabolic markers and reduced biological age, though long-term effects on lifespan remain unknown.
Stem cell exhaustion and tissue repair
Stem cell exhaustion impairs tissue regeneration and repair, contributing to sarcopenia, immune senescence, and delayed wound healing. Resistance training and adequate protein intake preserve muscle stem cell function. Maintaining vitamin D and testosterone levels supports stem cell activity and tissue maintenance. The decline in regenerative capacity is not fixed; it responds to environmental inputs.
What drives compression or expansion of morbidity
Whether you compress or expand morbidity depends on the balance between factors that accelerate disease onset and those that delay it. The inputs are behavioral, metabolic, and environmental.
Physical activity and cardiorespiratory fitness
Aerobic exercise delays the onset of cardiovascular disease, type 2 diabetes, dementia, and all-cause mortality more effectively than it extends maximum lifespan. VO2 max, a measure of cardiorespiratory fitness, is one of the strongest predictors of longevity and functional independence. Each 1-MET increase in VO2 max is associated with a 10-15% reduction in mortality risk.
Exercise stimulates mitochondrial biogenesis, improves insulin sensitivity, reduces systemic inflammation, and enhances cerebral blood flow. Resistance training preserves muscle mass and bone density, delaying sarcopenia and frailty. The effect is dose-dependent: higher levels of physical activity are associated with greater compression.
Metabolic health and glycemic control
Chronic hyperglycemia and insulin resistance accelerate vascular damage, neurodegeneration, and kidney disease. Maintaining fasting glucose below 90 mg/dL, HbA1c below 5.5%, and fasting insulin below 5 µIU/mL delays the onset of metabolic disease. Postprandial glucose spikes drive glycation and oxidative stress, contributing to advanced glycation end-product (AGE) accumulation in tissues. Dietary patterns that minimize glycemic variability, such as low-glycemic-index diets and time-restricted eating, reduce metabolic stress and delay disease onset.
Body composition and visceral adiposity
Visceral fat is metabolically active, secreting pro-inflammatory cytokines and adipokines that drive insulin resistance, dyslipidemia, and cardiovascular disease. Lean muscle mass, by contrast, is protective. Muscle acts as a metabolic sink for glucose, improves insulin sensitivity, and secretes myokines that reduce inflammation. Sarcopenia, the loss of muscle mass with age, is a strong predictor of disability and mortality. Maintaining muscle through resistance training and adequate protein intake (1.2-1.6 g/kg/day) delays functional decline.
Cardiovascular risk factors
Elevated apolipoprotein B (ApoB), lipoprotein(a), and blood pressure accelerate atherosclerosis and increase the risk of myocardial infarction and stroke. These events compress healthspan by introducing sudden disability. Controlling these risk factors through diet, exercise, and pharmacotherapy when indicated delays vascular disease onset. High-sensitivity C-reactive protein (hsCRP) reflects systemic inflammation and predicts cardiovascular events independently of cholesterol levels.
Sleep and circadian rhythm
Chronic sleep deprivation accelerates epigenetic aging, impairs glucose metabolism, and increases cardiovascular risk. Deep sleep is when growth hormone is secreted and the glymphatic system clears neurotoxic waste from the brain. Circadian rhythm disruption, from shift work or irregular sleep schedules, is associated with metabolic syndrome, cancer, and accelerated aging. Consistent sleep duration of 7-8 hours per night and alignment with natural light-dark cycles support compression.
Chronic stress and cortisol dysregulation
Chronic activation of the hypothalamic-pituitary-adrenal (HPA) axis elevates cortisol, which is catabolic to muscle and bone, suppresses immune function, and accelerates telomere shortening. Stress-induced inflammation drives multiple aging pathways. Interventions that modulate the stress response, such as mindfulness practices, social connection, and physical activity, reduce allostatic load and delay disease onset.
Why compression varies between individuals
Not everyone compresses morbidity to the same degree. The same interventions produce different outcomes depending on genetic background, baseline health, and cumulative exposures.
Certain genetic variants are associated with exceptional longevity and delayed disease onset. APOE genotype influences Alzheimer's risk and cardiovascular disease. APOE4 carriers have higher risk, while APOE2 is protective. FOXO3 variants are overrepresented in centenarians and are associated with delayed onset of age-related diseases. Variants in DNA repair genes, such as those involved in telomere maintenance, influence the rate of genomic instability. Genetics account for approximately 20-30% of lifespan variation, but a larger proportion of healthspan variation.
Biological age, as measured by epigenetic clocks, diverges from chronological age. Some individuals age faster, others slower. DunedinPACE measures the pace of aging and predicts functional decline, cognitive impairment, and mortality. Interventions that slow epigenetic aging, such as sustained improvements in diet, exercise, and metabolic health, are associated with compression. The rate of epigenetic aging is modifiable, but baseline pace varies.
Individuals vary in insulin sensitivity, mitochondrial efficiency, and substrate utilization. Some people maintain excellent glucose control despite high carbohydrate intake; others develop insulin resistance early. Metabolic flexibility, the ability to switch between fat and glucose oxidation, declines with age but varies widely. Those who maintain metabolic flexibility delay the onset of type 2 diabetes and cardiovascular disease.
Centenarians have distinct microbiome profiles characterized by higher diversity, greater abundance of butyrate-producing bacteria, and lower levels of pro-inflammatory species. The microbiome influences systemic inflammation, immune function, and metabolic health. Dysbiosis, characterized by low diversity and overgrowth of pathogenic species, accelerates aging. Diet, particularly fiber intake, shapes microbiome composition and influences compression potential.
Sex hormones modulate aging rate. Estrogen is protective against cardiovascular disease and bone loss; menopause accelerates both. Testosterone supports muscle mass, bone density, and metabolic health; age-related decline in testosterone contributes to sarcopenia and frailty. Growth hormone and IGF-1 decline with age, reducing tissue repair capacity. Hormonal transitions, such as menopause and andropause, accelerate specific aging pathways and influence the degree of compression achieved.
Lifetime exposure to environmental toxins, chronic infections, early life adversity, and socioeconomic stress accumulates as allostatic load. Higher allostatic load is associated with accelerated biological aging and earlier onset of chronic disease. Individuals with lower cumulative stress burden and fewer adverse exposures achieve greater compression.
What the evidence actually shows
The compression of morbidity hypothesis has been tested in multiple population studies over the past four decades. The evidence is mixed, but several patterns have emerged.
Longitudinal studies of older adults show that disability rates have declined in some cohorts, even as lifespan has increased. Data from the National Long Term Care Survey and the Health and Retirement Study indicate that the prevalence of disability among older Americans decreased between 1982 and 2004, consistent with compression. However, more recent data suggest that these gains have stalled or reversed in some populations, particularly among younger cohorts with high rates of obesity and metabolic disease.
The CALERIE trial, the first randomized controlled trial of caloric restriction in humans, demonstrated that modest caloric restriction (approximately 12% reduction) improved metabolic markers, reduced biological age as measured by epigenetic clocks, and decreased markers of inflammation and oxidative stress. These findings support the mechanistic basis for compression, though the trial was not powered to assess long-term effects on disease onset or mortality.
Studies of highly active older adults, such as the Stanford Running Study, show that regular vigorous exercise delays the onset of disability by approximately 16 years compared to sedentary controls. Runners maintained functional independence longer, and when disability did occur, it was compressed into a shorter period before death. This is one of the clearest examples of compression in a human cohort.
Epigenetic clock studies show that interventions targeting diet, exercise, and metabolic health can slow or reverse biological aging. A 2021 study using the DunedinPACE clock found that individuals with slower rates of biological aging had delayed onset of chronic disease and maintained functional capacity longer. However, whether slowing epigenetic aging translates to extended lifespan or simply compressed morbidity remains an open question.
The evidence from model organisms is more consistent. Caloric restriction, rapamycin (an mTOR inhibitor), and senolytic drugs extend healthspan more than lifespan in mice, rats, and other species. These interventions delay the onset of age-related pathologies and compress the period of functional decline. Whether these findings translate to humans is uncertain, but the mechanistic pathways are conserved.
Not all populations show compression. In some cohorts, particularly those with high rates of obesity, diabetes, and sedentary behavior, morbidity is expanding. Life expectancy is increasing, but so is the number of years spent with chronic disease. This divergence suggests that compression is not automatic; it requires active prevention.
Measuring your own trajectory
Compression of morbidity is not an abstract concept. It's a measurable outcome that depends on the trajectory of specific biomarkers over time. Tracking these markers longitudinally gives you a real signal on whether you're compressing or expanding morbidity.
Metabolic markers are foundational:
- Fasting glucose, fasting insulin, HbA1c, and the triglyceride-glucose index reflect insulin sensitivity and glycemic control.
- Rising trends indicate accelerating metabolic dysfunction and earlier onset of diabetes and cardiovascular disease.
- Stable or improving trends delay disease onset.
Cardiovascular markers predict vascular aging:
- ApoB reflects the number of atherogenic particles and is a stronger predictor of cardiovascular events than LDL cholesterol.
- Lipoprotein(a) is genetically determined but modifiable through lifestyle and pharmacotherapy.
- hsCRP reflects systemic inflammation and predicts cardiovascular risk independently of cholesterol.
- Homocysteine is a marker of methylation capacity and vascular health.
Inflammatory markers reflect the rate of inflammaging. hsCRP, erythrocyte sedimentation rate (ESR), and composite indices like the systemic immune-inflammation index (SII) capture chronic low-grade inflammation. Elevated inflammatory markers predict earlier onset of cardiovascular disease, dementia, and frailty.
Hormonal markers influence aging rate:
- IGF-1 reflects growth hormone status and tissue repair capacity.
- Testosterone and estradiol support muscle mass, bone density, and metabolic health.
- Cortisol reflects HPA axis function and chronic stress burden.
- Thyroid function influences metabolic rate and energy production.
Nutrient status markers support cellular function. Vitamin D, magnesium, vitamin B12, and folate are cofactors in methylation, DNA repair, and mitochondrial function. Deficiencies accelerate aging.
Body composition, measured by DEXA scan, tracks lean mass and visceral fat. Declining muscle mass and rising visceral fat predict earlier onset of metabolic disease and functional decline. Grip strength is a simple functional marker that predicts mortality and disability risk. A single measurement is a snapshot. A series of measurements over time is a trajectory. The rate of change matters more than any single value.
Building a compression strategy with comprehensive data
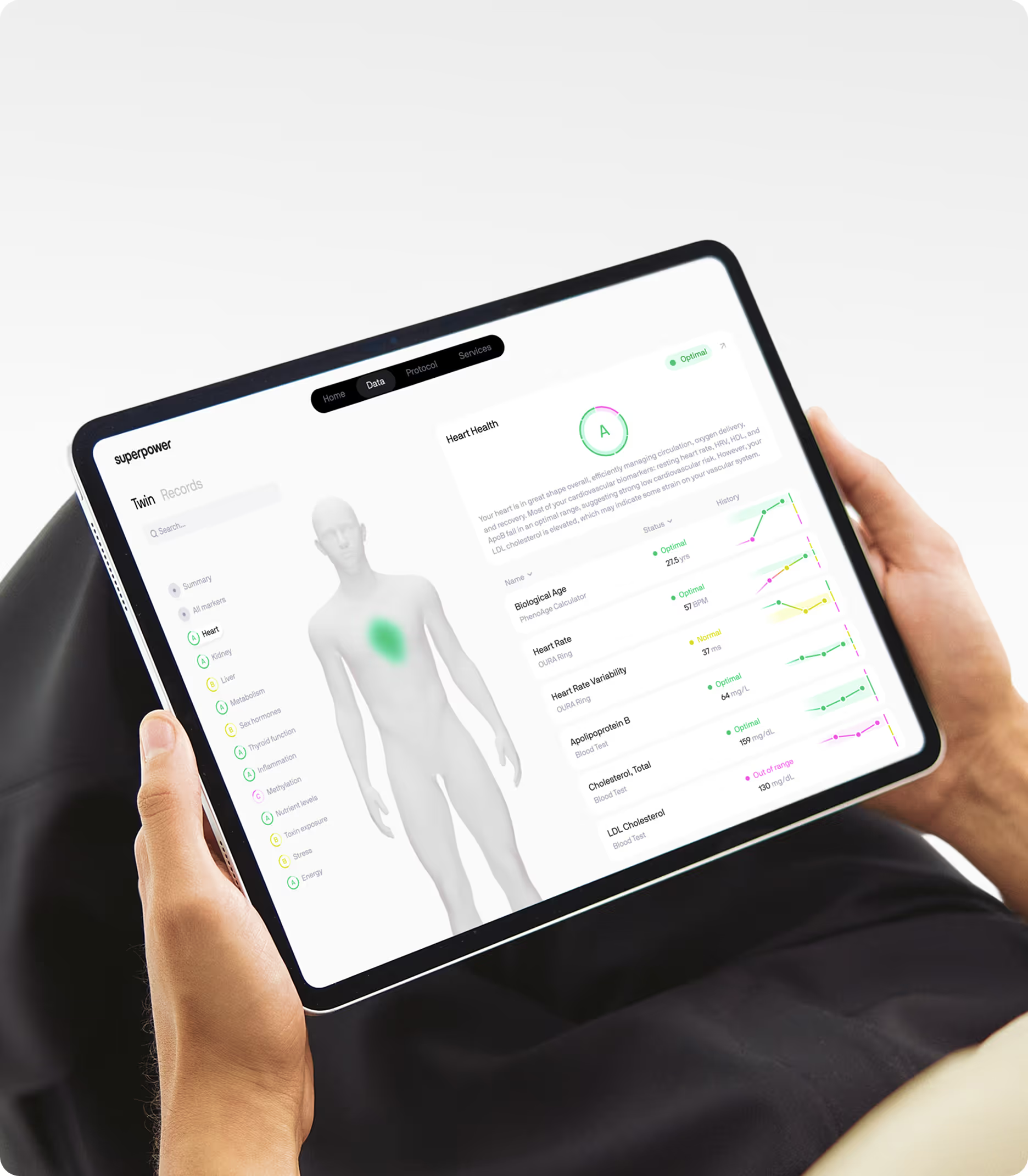
Compression of morbidity is not a passive outcome. It requires deliberate intervention, sustained over years, guided by longitudinal data. If you want to know whether you're compressing or expanding morbidity, you need to track the markers that predict disease onset, not just wait for symptoms to appear. Superpower's 100+ biomarker panel covers the metabolic, cardiovascular, inflammatory, and hormonal markers most relevant to aging rate and disease trajectory. Tracking these markers over time gives you the data to adjust course before chronic disease takes hold. Compression is not about living forever. It's about staying functional, independent, and healthy until the end.



FAQs
Compression of morbidity is the hypothesis introduced by Stanford physician James Fries in 1980. It proposes that the period of illness and functional decline at the end of life can be shortened — not extended — even as total lifespan increases. This happens when the onset of chronic disease is postponed more rapidly than mortality is delayed. Instead of spending decades managing multiple conditions, the goal is to remain functionally healthy until a brief window of decline just before death.
The Stanford Running Study showed that regular vigorous exercise delays the onset of disability by approximately 16 years compared to sedentary controls. Aerobic exercise delays cardiovascular disease, type 2 diabetes, and dementia more effectively than it extends maximum lifespan. Each 1-MET increase in VO2 max is associated with a 10 to 15 percent reduction in mortality risk. Exercise achieves compression by stimulating mitochondrial biogenesis, improving insulin sensitivity, reducing systemic inflammation, and preserving muscle mass and bone density.
Fasting glucose, fasting insulin, HbA1c, and the triglyceride-glucose index reflect insulin sensitivity and glycemic control. Rising trends in these markers signal accelerating metabolic dysfunction and earlier onset of diabetes and cardiovascular disease. ApoB reflects atherogenic particle burden and is a stronger cardiovascular predictor than LDL. hsCRP captures systemic inflammation. Tracking these markers longitudinally — not in a single snapshot — reveals whether disease onset is being delayed or accelerated relative to the calendar.
Compression potential varies because genetics, baseline metabolic health, and cumulative exposures all modulate how interventions work. APOE genotype, FOXO3 variants, and DNA repair gene polymorphisms set different biological baselines. Metabolic flexibility — the ability to switch between fat and glucose oxidation — varies widely and determines how quickly dietary changes shift biomarkers. Microbiome composition, hormonal milieu, and lifetime allostatic load further shape individual responses. Genetics account for roughly 20 to 30 percent of lifespan variation, but influence a larger share of healthspan variation.
The CALERIE trial was the first randomized controlled trial of caloric restriction in humans. A modest caloric reduction of approximately 12 percent improved metabolic markers, reduced biological age as measured by epigenetic clocks, and decreased markers of inflammation and oxidative stress. These findings support the mechanistic basis for compression by demonstrating that mTOR inhibition, AMPK activation, and enhanced autophagy from caloric moderation can slow biological aging in humans, though the trial was not designed to measure long-term effects on disease onset or mortality.
Senescent cells — those that have stopped dividing but have not died — secrete pro-inflammatory cytokines and adipokines through a process called the senescence-associated secretory phenotype. This chronic inflammatory signaling drives insulin resistance, atherosclerosis, and neurodegeneration. When the immune system loses its ability to clear senescent cells efficiently, their accumulation accelerates. In animal models, clearing senescent cells with senolytics extends healthspan more than lifespan, a pattern consistent with compression — disease onset is delayed without proportionally extending total lifespan.
References
- Centers for Disease Control and Prevention. (n.d.). About Chronic Diseases. https://cdc.gov/chronic-disease/about/index.html
- National Institute on Aging. (n.d.). What Do We Know About Healthy Aging?. https://nia.nih.gov/health/healthy-aging/what-do-we-know-about-healthy-aging
- Fries, J. F., Bruce, B., & Chakravarty, E. (2011). Compression of morbidity 1980-2011: a focused review of paradigms and progress. Journal of aging research, 2011, 261702. https://doi.org/10.4061/2011/261702
- Fries, J. F. (1980). Aging, natural death, and the compression of morbidity. The New England journal of medicine, 303(3), 130-5. https://doi.org/10.1056/NEJM198007173030304
- Geyer, S., & Eberhard, S. (2022). Compression and Expansion of Morbidity—Secular Trends Among Cohorts of the Same Age. Deutsches Arzteblatt international, 119(47), 810-815. https://doi.org/10.3238/arztebl.m2022.0324

























.avif)
.svg)
.svg)


